Ground-state relaxation and finite temperatures
This web page can be downloaded as notebook: relaxation.ipynb (Jupyter)
or relaxation.md (Markdown)
Introduction
In imaginary time, 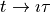 , the Schrödinger equation becomes
(we use atomic units throughout the text)
, the Schrödinger equation becomes
(we use atomic units throughout the text)
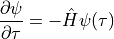
with the formal solution of the time evolution operator
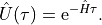
There are two main applications for such a time evolution:
If you apply this time evolution operator to some wave function, the norm decays. The decay is faster for high-energy contributions, so that after sufficient propagation time, the wave function is dominated by the ground state. Hence, imaginary-time propagation is an easy way to get the ground state of a system (and also some excited states).
The time evolution operator has the shape of a density operator for the system at finite temperature. Hence, you can use imaginary-time propagation to prepare a system at a finite temperature. There are actually different techniques to do so, this is detailed in Representing states at finite temperature.
Relaxing wave functions
Calculating the ground state
At its heart, the relaxation solver is just a modified version [1]
of the wavepacket.solver.ChebychevSolver,
whose use is detailed in Using Chebychev solvers.
Most remarks in the reference apply here as well, in particular the spectrum guess and operator truncation.
We take the harmonic oscillator example of Using Chebychev solvers,
import numpy as np
import wavepacket as wp
grid = wp.grid.Grid(wp.grid.PlaneWaveDof(-10, 10, 128))
kinetic = wp.operator.CartesianKineticEnergy(grid, 0, mass=1, cutoff=35)
potential = wp.operator.Potential1D(grid, 0, lambda x: 0.5 * x ** 2, cutoff = 35)
hamiltonian = kinetic + potential
solver = wp.solver.RelaxationSolver(hamiltonian, 1.5, (0, 70))
In contrast to the real-time evolution, however, we always apply the Hamiltonian from the left, also for density operators. Hence, the solver takes the Hamiltonian as argument, not a Schrödinger equation or Liouvillian.
Because of the exponential dynamics, most details of the initial state are irrelevant, as long as it has some overlap with the true ground state. In practice, you just take something simple like a Gaussian somewhere around the potential minimum and let the solver optimize the state.
Typically, we do not care deeply about intermediate results, only about the final ground state, and only use the intermediate results to monitor convergence. Note, though, that the result needs to be normalized before further use!
def relax(solver, psi0):
for t, psi in solver.propagate(psi0, t0=0, num_steps=5):
psi_normalized = wp.normalize(psi)
energy = wp.expectation_value(hamiltonian, psi_normalized).real
energy2 = wp.expectation_value(hamiltonian*hamiltonian, psi_normalized).real
print(f"t = {t}: E = {energy}, dE^2 = {energy2 - energy**2:.4}, |psi|^2 = {wp.trace(psi):.4}")
return psi
# to demonstrate convergence, let us take a Gaussian that is too wide (rms=1 would be exact)
psi0 = wp.builder.product_wave_function(grid, wp.special.Gaussian(rms=2))
relax(solver, psi0);
t = 0: E = 1.0624999933916848, dE^2 = 1.758, |psi|^2 = 1.0
t = 1.5: E = 0.5008931477847255, dE^2 = 0.001788, |psi|^2 = 0.1786
t = 3.0: E = 0.5000022119213396, dE^2 = 4.424e-06, |psi|^2 = 0.03983
t = 4.5: E = 0.5000000054827928, dE^2 = 1.097e-08, |psi|^2 = 0.008887
t = 6.0: E = 0.5000000000135902, dE^2 = 2.718e-11, |psi|^2 = 0.001983
t = 7.5: E = 0.5000000000000335, dE^2 = 6.75e-14, |psi|^2 = 0.0004425
Note how the energy uncertainties decay exponentially by about a factor of five for each time step. In the example here, we are basically converged after about three steps, but the relaxation is usually the least expensive part of the calculation, so you can afford to spend a few more CPU cycles on a converged solution.
Beware that the initial state must have overlap with the ground state!
This most often fails because the symmetry is incorrect.
In our harmonic oscillator example, the ground state has even parity, 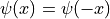 .
Let us check what happens if we choose an initial state with odd parity, for example a sign function:
.
Let us check what happens if we choose an initial state with odd parity, for example a sign function:
psi0 = wp.builder.product_wave_function(grid, lambda x: np.sign(x))
relax(solver, psi0);
t = 0: E = 16.421638040536198, dE^2 = 190.7, |psi|^2 = 1.0
t = 1.5: E = 1.5008494577695273, dE^2 = 0.001718, |psi|^2 = 0.002518
t = 3.0: E = 1.5000020817512834, dE^2 = 4.164e-06, |psi|^2 = 2.796e-05
t = 4.5: E = 1.5000000051599987, dE^2 = 1.032e-08, |psi|^2 = 3.106e-07
t = 6.0: E = 1.5000000000127902, dE^2 = 2.558e-11, |psi|^2 = 3.45e-09
t = 7.5: E = 1.5000000000000313, dE^2 = 6.35e-14, |psi|^2 = 3.833e-11
Neat, isn’t it? We converged, but to the first excited state, because the ground state has exactly zero overlap with an odd function. In general, you should, however, not rely on this behavior. If the wave function has or acquires even the smallest asymmetry, for example through finite-precision arithmetics, you will eventually converge to the ground state, albeit slowly.
However, you can exploit this behavior in a different scenario. The time required for convergence is given by the smallest excitation energy, because that state’s contribution decays second-slowest after the ground-state contribution. In some systems, this excitation energy is small, leading to rather slow convergence. A prototypical example is the double-well potential, where low-energy states always come in pairs with small energy gaps within the pair. In most such cases, however, the two close-lying energy eigenstates differ by their symmetry. By selecting an initial state with the same symmetry as the ground state, you can already suppress the unwanted state from the start, leading to faster convergence.
Calculating excited states
With a bit more machinery, we can also calculate excited energy eigenstates. Imaginary-time propagation works because contributions from excited energy eigenstates decay faster than the ground-state contribution.
Now we do the following:
We calculate the ground state with imaginary-time propagation.
We run another imaginary-time propagation, and repeatedly remove the ground state component.
What happens? Well, the resulting wave function is dominated by its slowly decaying ground state component. If we remove it, we are left with the second-slowest decaying component, which is the first excited state. In this fashion, we can calculate successive excited states. Let us write the code before a discussion:
def print_data(state, n, step):
energy = wp.expectation_value(hamiltonian, state).real
energy2 = wp.expectation_value(hamiltonian*hamiltonian, state).real
print(f"step {step}: E_{n} = {energy}, dE^2 = {energy2 - energy**2:.4}")
def relax_v2(solver, found_states):
psi = wp.builder.product_wave_function(grid, wp.special.Gaussian(x=1, rms=2))
print_data(psi, len(found_states), 0)
for step in range(5):
psi = solver.step(psi, 0)
psi = wp.orthonormalize(found_states + [psi])[-1]
print_data(psi, len(found_states), step+1)
print("----------------------")
return psi
ground = relax_v2(solver, [])
exc1 = relax_v2(solver, [ground])
exc2 = relax_v2(solver, [ground, exc1])
# and so on
step 0: E_0 = 1.5624997861628531, dE^2 = 3.758
step 1: E_0 = 0.5051251772050889, dE^2 = 0.006281
step 2: E_0 = 0.5002011032583435, dE^2 = 0.0002039
step 3: E_0 = 0.5000098797293601, dE^2 = 9.887e-06
step 4: E_0 = 0.5000004915542029, dE^2 = 4.916e-07
step 5: E_0 = 0.5000000244722282, dE^2 = 2.447e-08
----------------------
step 0: E_1 = 1.5624997861628531, dE^2 = 3.758
step 1: E_1 = 1.629042635538908, dE^2 = 0.1156
step 2: E_1 = 1.507154910369327, dE^2 = 0.007111
step 3: E_1 = 1.5003582896725427, dE^2 = 0.0003582
step 4: E_1 = 1.500017820158577, dE^2 = 1.787e-05
step 5: E_1 = 1.5000008639725828, dE^2 = 9.129e-07
----------------------
step 0: E_2 = 1.5624997861628531, dE^2 = 3.758
step 1: E_2 = 2.5118036169340523, dE^2 = 0.01343
step 2: E_2 = 2.500510813621739, dE^2 = 0.0005165
step 3: E_2 = 2.5000244007784573, dE^2 = 2.619e-05
step 4: E_2 = 2.500000370162362, dE^2 = 2.147e-06
step 5: E_2 = 2.499999174216421, dE^2 = 9.511e-07
----------------------
A few notes about the code:
I have reduced the number of steps to 5 to compress the output, and because the results are already sufficiently converged.
Note that the initial state is now a shifted Gaussian. This is a deliberate choice. Because the eigenstates of the harmonic oscillator toggle between even and odd parity, we need an asymmetric initial state that contains contributions with both parities. If I had chosen an even or odd function as initial state, I would only get excited states of that parity.
The code exploits a semi-internal detail of the orthonormalization function; it normalizes the first entry, then orthonormalizes the second etc., so that the last entry is our wave function with the other components removed.
We manipulate the state while we evolve it in time. Therefore we can no longer
propagate()in one go, but must explicitly step through the solution.
In theory, you can now go on to get arbitrary excited states with this technique. In practice, you need a full propagation for each subsequent excited state, so this approach becomes very expensive very quickly. Also, errors may add up in larger systems. Any error in the ground state, because you did not propagate long enough, will lead to an incorrect orthogonalization of the first excited state and so on. The recovered states hence become more and more incorrect. Unfortunately, it is difficult to quantify this latter error even with a lot of hand waving, and at least this harmonic oscillator example seems pretty robust against this issue.
In conclusion, you may only want to recover a few excited states of the Hamiltonian and monitor the convergence carefully.
Propagating density operators in imaginary time
The calculation of the density operator for finite temperatures is derived in a roundabout way. The Liouville-von-Neumann (LvNe) equation

can be shown, in analogy to ordinary differential equations and Schrödinger equations, to have the solution
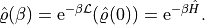
So we just set up the LvNe, and evolve the unit density in time until the requested inverse temperature. The step should be chosen to fit the requested inverse temperature(s) and keep the alpha value in a reasonable range. as an example, let us calculate the partition sum of our harmonic oscillator.
delta_beta = 1.0
solver = wp.solver.RelaxationSolver(hamiltonian, delta_beta, (0, 70))
rho = wp.builder.unit_density(grid)
for step in range(5):
rho = solver.step(rho, 0)
print(f"beta = {delta_beta * (step+1)}, Z = {wp.trace(rho):.4}")
beta = 1.0, Z = 0.9595
beta = 2.0, Z = 0.4255
beta = 3.0, Z = 0.2348
beta = 4.0, Z = 0.1379
beta = 5.0, Z = 0.08264
Because imaginary-time propagation only makes sense for a particular LvNE, you supply again the Hamiltonian directly, not some Liouvillian.
Side note: because the Liouvillian has the same spectrum as the Hamiltonian, we can use the same RelaxationSolver for density operators and wave functions, in contrast to the real-time ChebychevSolver.